
Innhold
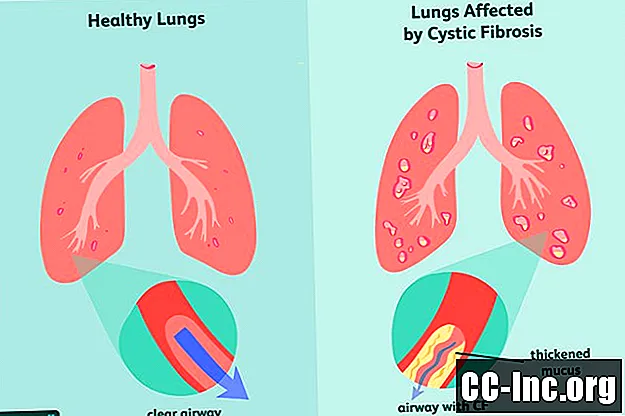
Cystisk fibrose (CF) er en arvelig, livstruende lidelse som skader lungene og fordøyelseskanalen. Det er forårsaket av et defekt gen som utløser produksjonen av tykkere slim som tetter luftveiene og blokkerer utskillelsen av fordøyelsesenzymer.Symptomene er progressive og ofte alvorlige, og de kan omfatte pusteproblemer, tilbakevendende lungeinfeksjoner, dårlig vekst, mannlig infertilitet og kronisk betennelse i bukspyttkjertelen, leveren, nyrene og hjertet.
CF kan diagnostiseres med blodprøver, genetisk screening og en prosedyre kjent som svettekloridtest.
Selv om det ikke finnes noen kur mot CF, finnes det behandlinger som kan forbedre lengden og kvaliteten på ens liv.
Disse inkluderer luftveisgodkjenningsteknikker, inhalerte antibiotika, slimfortynnere, bukspyttkjertelenzymer, et kaloririkt kosthold og nyere generasjons medisiner kjent som CFTR-modulatorer. I alvorlige tilfeller kan det være behov for en lungetransplantasjon.
Cystisk fibrose symptomer
Som en genetisk lidelse er cystisk fibrose noe du er født med. Det kan eller ikke kan ha symptomer på fødselen og kan ofte ta måneder eller til og med år før tegn på sykdom dukker opp. På den tiden kan lungene og fordøyelseskanalen allerede ha opplevd skade som ikke kan angres.
De vanligste tidlige tegn og symptomer på CF inkluderer:
- Blokkering av babyens første avføring (mekonium)
- Salt smakende hud
- En kronisk hoste, tungpustethet eller farget sputum
- Løs, fettete og typisk illeluktende avføring
- Lunginfeksjon, ofte tilbakevendende
- Dårlig vekst og svikt i å trives
Med mindre disse symptomene kan kontrolleres, kan stress på lungene (og manglende evne til å gå opp i vekt) ha en kumulativ effekt, som påvirker flere organer og øker risikoen for sykdomskomplikasjon.
Noen av de mer karakteristiske komplikasjonene inkluderer:
- Forsinket pubertet
- Bronkiektase (kronisk fortykning av lungeveggene)
- Vekttap
- Pankreatitt (betennelse i bukspyttkjertelen)
- Mannlig infertilitet
- Lungehypertensjon (høyt blodtrykk i lungen)
- Gallestein
- Cystisk fibrose-relatert diabetes
- Cor pulmonale (høyresidig hjertesvikt)
- Skrumplever (funksjonell arrdannelse i leveren)
Fordi CF forårsaker progressiv skade på celler og vev, vil eventuelle skader forårsaket av lungene og andre organer i stor grad være irreversible. Døden vil oftest være et resultat av respirasjonssvikt, etterfulgt av hjertesvikt og leversvikt.
Symptomer på cystisk fibrose
Fører til
Cystisk fibrose er forårsaket av mutasjonen av det cystiske fibrose transmembrane reseptor (CFTR) genet, som er ansvarlig for å produsere CFTR proteinet. Dette er proteinet kroppen trenger for å regulere strømmen av salt og vann inn og ut av celler . Hvis proteinet er deformert eller mangelfullt, kan det forårsake dehydrering på overflaten av en celle, noe som fører til fortykning av det omkringliggende slimet.
CF er en autosomal recessiv lidelse, noe som betyr at du må arve CFTR-mutasjonen fra både mor og far for å få sykdommen. Hvis du bare arver ett defekt gen, vil du ikke ha CF, men i stedet være en bærer av det muterte genet.
Du kan arve sykdommen hvis hver av foreldrene dine enten har en CFTR-mutasjon eller CF selv. Hvis begge foreldrene er bærere, vil du ha:
- 25 prosent sjanse for å få CF
- 50 prosent sjanse for å være transportør
- 25 prosent sjanse for å bli upåvirket
På den annen side, hvis en av foreldrene dine er transportør og den andre har CF, har du en 50/50 sjanse for å enten ha CF eller være transportør.
Cystisk fibrose er en av de vanligste genetiske sykdommene, og rammer omtrent en av hver 2500 babyer født i USA.
Det er mest vanlig blant kaukasiske og latinamerikanere, og forekommer sjeldnere hos mennesker av afrikansk eller asiatisk avstamning.
Risikofaktorer for cystisk fibroseDiagnose
Det er noen få tester som brukes til å diagnostisere cystisk fibrose. De fungerer enten ved direkte å oppdage CFTR-mutasjonen eller indirekte måle biologiske endringer i samsvar med sykdommen. Diagnosemetoden kan variere under graviditet, når babyen blir født, eller når som helst etterpå.
Diskusjonsveiledning for cystisk fibrose doktor
Få vår utskrivbare guide for din neste legeavtale for å hjelpe deg med å stille de riktige spørsmålene.

Av de to standardtestene som ofte brukes til å diagnostisere CF:
- Svetteklorid testing, også kjent som svettest, måler kloridmengden på huden. Fordi CF forstyrrer overføringen av salt til og fra celler, vil det være en opphopning av salt i svette.
- Genetisk CFTR-testing brukes til å oppdage de vanligste mutasjonene av CFTR-mutasjonen. Mens det er over 2000 CFTR-mutasjoner som er kjent for å forårsake cystisk fibrose, representerer de 23 som er inkludert i standardpanelet de mest sannsynlige mistenkte.
Under graviditet kan CFTR genetisk test brukes til å teste væsker oppnådd gjennom fostervannsprøve eller celler ekstrahert via chorionic villus sampling (CVS).
Nyfødt screening er også standard brukt til å diagnostisere CF og er i dag mandat i alle 50 delstater og District of Columbia. Hva dette innebærer vil variere avhengig av hvor i USA du bor. Hvis de nyfødte screeningresultatene er positive, vil en svettetest brukes til å bekrefte diagnosen.
Hvordan diagnostiseres cystisk fibroseBehandling
Selv om det ikke finnes noen kur mot cystisk fibrose, har fremskritt i behandlingen forlenget levetiden til de som lever med sykdommen.
Målet med CF-behandling er firdoblet: å forhindre infeksjoner, beholde lungefunksjonen, normalisere fordøyelsen og senke utviklingen av sykdommen.
Blant de terapeutiske verktøyene som brukes til å håndtere CF:
- Teknikker for klarering av luftveier (ACTs) utføres for å løsne og drive ut akkumulert slim fra lungene. Teknikker inkluderer huff hoste, perkusjon av brystet eller svingning i brystveggen.
- Et høyt fettinnhold med høyt kaloriinnhold brukes til å kompensere for malabsorpsjon av fett, proteiner og næringsstoffer i tarmene.
- Bukspyttkjertelenzymtilskudd brukes til å styrke fordøyelsesenzymer som bukspyttkjertelen ikke kan produsere på grunn av overdreven opphopning av slim.
- Antibiotika tas daglig for å forhindre bakterielle lungeinfeksjoner.
- Mukolytika-medisiner som brukes til å tynne slim før ACT-kan brukes.
- CFTR-modulatorer er en ny klasse medikamenter som kan rette opp visse feil i CFTR-proteinet og gjenopprette deres regulatoriske funksjon.
- Oksygenbehandling kan brukes under akutte episoder når pusten din har blitt sterkt svekket.
- Enteral ernæring, også kjent som tubefôring, kan brukes hvis du ikke tåler vekt gjennom normal ernæring.
- Lungetransplantasjon blir vurdert når lungene dine ikke lenger kan støtte overlevelse uten mekanisk ventilasjon.
Mestring
I 1938, da cystisk fibrose først ble klassifisert som en sykdom, levde barn sjelden utover sitt første leveår. På 1980-tallet kunne man forvente å leve så lenge som 20 til 25 år. I dag har bildet endret seg fullstendig med mennesker som lever langt inn i 40- og 50-årene hvis behandlingen startes tidlig og følges.
Dette er ikke et tegn på at CF er mindre alvorlig enn det noen gang har vært. Det er en livsendrende begivenhet som krever flid og konsistens for ikke bare å takle sykdommen, men også leve den høyeste mulige livsstandard.
For dette formål må du normalisere CF i livet ditt ved å etablere rutiner og praksis for å unngå oppturer og nedturer som kan forårsake stress og øke funksjonshemming. Blant hensynene må du:
- Administrer ernæringen din. Mennesker med CF trenger ofte dobbelt så mange kalorier daglig som andre mennesker gjør.
- Trene regelmessig. Treningsrutiner bør ideelt sett innebære minst 20 til 30 minutters aerob aktivitet tre ganger per uke. Finn noe hyggelig du kan gjøre i livet.
- Hold deg godt hydrert. Dette gjør at lungene og tarmene fungerer som de skal. Avhengig av alder, bør du drikke ikke mindre enn seks til åtte høye glass vann per dag.
- Utfør luftveisklarering riktig. Etter hvert som helsebehovene dine endres, kan også typene klareringsverktøy du trenger. Snakk med lungelege eller fysioterapeut hvis du ikke oppnår de resultatene du burde.
- Søk støtte. I tillegg til venner og familie, kan du kontakte nærmeste kapittel i Cystic Fibrosis Foundation (CFF) for å koble til et støttenettverk i ditt område.
- Søk økonomisk hjelp. CFF tilbyr tjenester som hjelper familier bedre å takle de høye kostnadene ved CF-behandling.
Et ord fra veldig bra
Mens screening av nyfødte har økt frekvensen av CF-diagnoser hos babyer dramatisk, blir over 25 prosent av diagnosene bare gjort i barndoms-, ungdoms- og tidlig voksenår.
Dette er problematisk fordi tidlig diagnose og behandling kan avverge mange av de mer alvorlige komplikasjonene av CF før alvorlig skade kan gjøres. Selv om behandlingen ikke kan stoppe eller reversere sykdommen, kan den sikre mange flere sykdomsfrie år.
For å oppnå dette er det viktig å kjenne til de tidlige symptomene på CF og snakke med legen din hvis du mistenker at barnet ditt kan ha sykdommen. Dette gjelder spesielt i stater som bare viser med IRT-blodprøver, noe som kan resultere i at så mange 5 prosent av barna opplever enten en forsinket diagnose eller et falskt negativt resultat, ifølge forskning fra University of Wisconsin School of Medicine and Public Health. .
Hvilke symptomer kan du forvente med cystisk fibrose?